|
Welcome
People
Research
Publications
Teaching
Join us
Contact
Impressum
|
Main research topics
The HMI group combines multiscale simulations including DFT-based electronic structure calculations, first-principles
molecular dynamics, all-atom classical molecular dynamics and coarse-grained dissipative particle dynamics
with experimental methods such as atomic force microscopy, circular dichroism spectroscopy
and x-ray microscopy to characterize and understand the behaviour of heterogeneous materials.
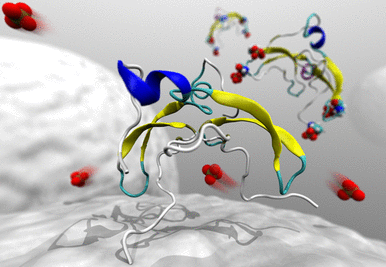
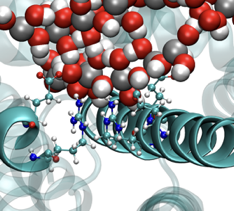
| Bio-hybrid interfaces
We perform atomic-level studies of proteins, nucleic acids, phospholipids and polysaccharides,
both as pure biological systems and interfaced with metals, oxides, carbon allotropes and
two-dimensional materials.
We quantify the strength of the interfacial interactions in terms of adhesion
forces and binding free energy and we reveal conformational changes that occur as a consequence
of such interactions. This allows us to rationalise and guide the design of novel functional
materials and coatings, and to understand the molecular mechanisms at the basis of important
biochemical and biological processes. Current research topics include biomineralization,
protein glycosylation, micro-RNA structure/function relationships, organic nanofibre formation.
|
|
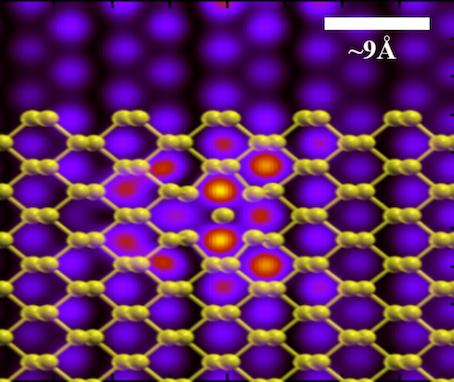
| Inorganic hybrids
Combining atomistic and coarse-grained simulations with AFM imaging and force spectroscopy
we study the behaviour of inorganic systems composed of heterogeneous phases such as nanoparticle
films in a gas or liquid atmosphere, oxide layers growing on functional substrates, mesoporous materials
or electrochemical interface systems. Applications comprise the prediction of transport properties in
functional two-dimensional materials, the behaviour of battery electrodes, the handling of nanoparticle
films for the fabrication of gas sensors and catalysts. Electronic-structure methods at the DFT level and
beyond are employed in combination with structural and electrochemical characterization experiments
performed by our collaboration partners.
|

|
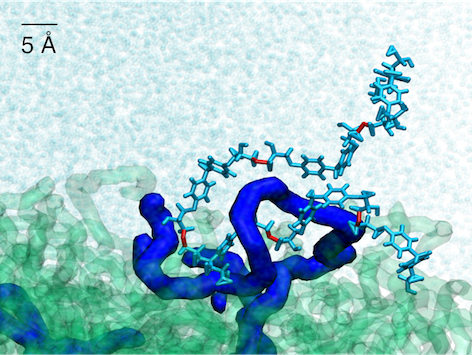
| Technical polymer interfaces
As an emerging research line in our group, we investigate the properties of polymeric
materials interfaces at the all-atom and coarse-grained levels.
This includes the engineering of technical polymers, for instance in the context of thermoplastic welding or
thermoplastic/thermoset co-curing, as well as natural polymers such as chitosan, fibrin and collagen,
for instance for the fabrication of tissue-engineering scaffolds and drug-delivery systems. We use
CD spectroscopy to study changes in the secondary structure of natural fibres upon assembling and
employ enhanced-sampling molecular dynamics methods to access the complex conformational phase space
of the systems.
|

|
|